Как рассчитать экспериментальную ошибку в химии
На чтение 1 мин. Просмотров 262 Опубликовано 05.06.2021
Ошибка – это мера точности значений в вашем эксперименте. Важно уметь вычислить экспериментальную ошибку, но есть несколько способов ее вычислить и выразить. Вот наиболее распространенные способы вычисления экспериментальной ошибки:
Содержание
- Формула ошибки
- Формула относительной ошибки
- Формула процента ошибки
- Пример расчета ошибки
Формула ошибки
В общем, ошибка – это разница между принятым или теоретическое значение и экспериментальное значение.
Ошибка = экспериментальное значение – известное значение
Формула относительной ошибки
Относительная ошибка = ошибка/известное значение
Формула процента ошибки
% Error = относительная ошибка x 100%
Пример расчета ошибки
Допустим, исследователь измеряет массу образца, который должен быть 5,51 грамм. Известно, что фактическая масса образца составляет 5,80 грамма. Рассчитайте погрешность измерения.
Экспериментальное значение = 5,51 грамма
Известное значение = 5,80 грамма
Ошибка = экспериментальное значение – известное значение
Ошибка = 5,51 г – 5,80 грамма
Ошибка = – 0,29 грамма
Относительная ошибка = ошибка/известное значение
Относительная ошибка = – 0,29 г/5,80 г
Относительная ошибка = – 0,050
% Error = относительная ошибка x 100%
% Error = – 0,050 x 100%
% Error = – 5,0%
Actually the % error is calculated as:
$frac{text{experimental} — text{actual}}{text{actual}}cdot100%$
That way
- if experimental < actual then the error is negative
- if experimental > actual then the error is positive
So my question is if the experimental is close enough to the actual that the subtraction would give zero significant figures, would then the percent error then be zero?
Not exactly zero…
EX: measured density of 0.997g/ml and actual density of 0.997171g/ml then, 0.997171-0.997=0.000171 which would be rounded to 0.000 due to sig. figs
Correct for subtraction you have used. Addition and subtraction round to the the last common significant decimal place of all the measurements. That would be the 8 in 10.98 in the following example.
So $10.9bar8 + 0.6754 + 0.4795 = 12.1349 approx 12.13 $
In actually you should have used the reverse
0.997 — 0.99717 = -0.000171
there are no significant figures so this would mean the calculated 0.0171% error would be zero correct.
Now this gets sticky. You could write that the error was 0% but that would indicate only one part in 100 precision. If you write 0.00% it isn’t entirely clear if that is two or three significant figures. But you have claimed that the hundredth place in the % is significant, which it is not.
Remember that the minimum nonzero error possible was +/- 0.001 which yields an error of 0.1%.
So the error would be best written as 0.0% indicating that the error is 0 to 1 part in a thousand.
Reference: Significant Figures at Wikipedia
I’ll leave the propagation of errors for a different exercise.
Actually the % error is calculated as:
$frac{text{experimental} — text{actual}}{text{actual}}cdot100%$
That way
- if experimental < actual then the error is negative
- if experimental > actual then the error is positive
So my question is if the experimental is close enough to the actual that the subtraction would give zero significant figures, would then the percent error then be zero?
Not exactly zero…
EX: measured density of 0.997g/ml and actual density of 0.997171g/ml then, 0.997171-0.997=0.000171 which would be rounded to 0.000 due to sig. figs
Correct for subtraction you have used. Addition and subtraction round to the the last common significant decimal place of all the measurements. That would be the 8 in 10.98 in the following example.
So $10.9bar8 + 0.6754 + 0.4795 = 12.1349 approx 12.13 $
In actually you should have used the reverse
0.997 — 0.99717 = -0.000171
there are no significant figures so this would mean the calculated 0.0171% error would be zero correct.
Now this gets sticky. You could write that the error was 0% but that would indicate only one part in 100 precision. If you write 0.00% it isn’t entirely clear if that is two or three significant figures. But you have claimed that the hundredth place in the % is significant, which it is not.
Remember that the minimum nonzero error possible was +/- 0.001 which yields an error of 0.1%.
So the error would be best written as 0.0% indicating that the error is 0 to 1 part in a thousand.
Reference: Significant Figures at Wikipedia
I’ll leave the propagation of errors for a different exercise.
![]()
Загрузить PDF
![]()
Загрузить PDF
Погрешность измерения, выраженная в процентах (далее процентная погрешность) — это разность между истинным и измеренным значением, деленная на истинное значение и умноженная на 100. Процентная погрешность позволяет представить, насколько (в процентах) измеренное значение отличается от истинного. Погрешность может быть вызвана ошибками в измерениях (неточными инструментами или человеческим фактором) или из-за округлением значений. При этом формула для вычисления процентной погрешности довольно простая.
-

1
Запишите формулу для вычисления процентной погрешности. Формула: [(|Измеренное значение — Истинное значение|) / Истинное значение] x 100. В эту формулу необходимо подставить два значения — измеренное и истинное.[1]
- Измеренное значение — это оценочное (приблизительное) значение; истинное значение — это точное значение.
- Например, если вы думаете, что в сумке лежат 9 апельсинов, но на самом деле их 10, число 9 — это приблизительное значение, а 10 — точное значение.
-

2
Вычтите точное значение из оценочного. В нашем примере вычтите 10 (точное значение) из 9 (оценочное значение): 9 — 10 = -1.[2]
- Эта разность характеризует различие между приблизительным и точным значениями, то есть насколько точное значение отличается от оценочного.
-

3
Найдите абсолютное значение этой разности. Так как в формулу нужно подставить абсолютное значение разности, знаком «минус» можно пренебречь. То есть в нашем примере -1 превратится в 1.[3]
- В нашем примере: 9 — 10 = -1. Абсолютное значение -1 записывается так: |-1| = 1.
- Если разность положительная, не меняйте ее. Например: 12 яблок (приблизительное значение) — 10 яблок (точное значение) = 2. Абсолютное значение 2: |2| = 2.
- В статистике абсолютное значение означает, что вас не интересует, в каком направлении отклоняется оценочное значение (слишком большое, то есть положительное, или слишком маленькое, то есть отрицательное). Вы просто хотите знать, на какую величину оценочное значение отличается от истинного.
-

4
Разделите найденную разность на абсолютную величину истинного значения. Сделайте это на калькуляторе или вручную. В нашем примере точное значение уже положительное, поэтому просто разделите 1 (полученная разность) на 10 (точное значение).[4]
- В нашем примере: 1/|10|= 1/10.
- В некоторых случаях точное значение может быть отрицательным числом. Если это так, знак «минус» можно проигнорировать (то есть работайте с абсолютной величиной точного значения).[5]
Реклама
-

1
Преобразуйте обычную дробь в десятичную. В проценты проще преобразовать десятичную дробь. В нашем примере 1/10 = 0,1. Более сложные вычисления выполните на калькуляторе.
- Если под рукой калькулятора нет, разделите числа в столбик, чтобы получить десятичную дробь. Как правило, достаточно 4–5 цифр после десятичной запятой, чтобы округлить дробь.
- При преобразовании обычной дроби в десятичную всегда делите положительное число на положительное число.
-

2
Умножьте полученную десятичную дробь на 100. В нашем примере умножьте 0,1 на 100, а затем к результату припишите символ «%». Так вы получите процентную погрешность.[6]
- В нашем примере: 0,1 x 100 = 10 %.
-

3
Проверьте результат, чтобы убедиться, что он правильный. Иногда замена знаков (положительный/отрицательный) и деление могут привести к незначительным ошибкам в расчетах. Поэтому лучше проверить ответ.
- В нашем примере необходимо убедиться, что оценочное значение (9 апельсинов) отличается от истинного значения (10 апельсинов) на 10 %. 10 % (10 % = 0,1) от 10 апельсинов равно 1 (0,1 × 10 = 1).
- 9 апельсинов + 1 = 10 апельсинов. Это подтверждает, что оценочное значение (9) действительно отличается от истинного значения (10) на 1 (то есть на 10 %).
Реклама
Советы
- Иногда измеренное (оценочное, приблизительное) значение называется экспериментальным, а истинное (точное) значение называется теоретическим. Обязательно используйте значение, с которым сравнивается данное значение, как точное значение.
- Так как в данном методе используются абсолютные величины приблизительных и точных значений, нет разницы, в каком порядке вычитать числа. Например,|8 — 4| = 4 и |4 — 8| = |-4|= 4. Результаты одинаковые!
Реклама
Об этой статье
Эту страницу просматривали 63 086 раз.
Была ли эта статья полезной?
Формула процентной ошибки (Содержание)
- Формула процентной ошибки
- Примеры формулы ошибки процента (с шаблоном Excel)
- Калькулятор формулы ошибки процента
Формула процентной ошибки
Формула процентной погрешности используется во многих научных отчетах и особенно в химии, формула используется для измерения разницы между измеренным или экспериментальным значением и истинным или точным значением. Цель формулы процента ошибки состоит в том, чтобы измерить, насколько измеренное значение отличается от точного значения. Вычисление формулы процентной ошибки включает абсолютную ошибку, которая просто в большинстве случаев обозначается в положительном значении. Обратите внимание, что для химии и других наук принято хранить отрицательное значение. Важна ли ошибка положительная или отрицательная. Формула процентной ошибки, близкая к нулю или равная нулю, означает, что пользователь очень близок к целевому значению, которое является положительным знаком для пользователя. Но для пользователя очень важно знать причину и обоснование причины ошибки, которая может возникнуть из-за различных причин, таких как ошибка, произошедшая во время эксперимента, или ошибка в его собственной оценке. Формула все о сравнении предположения или оценки с точным значением.
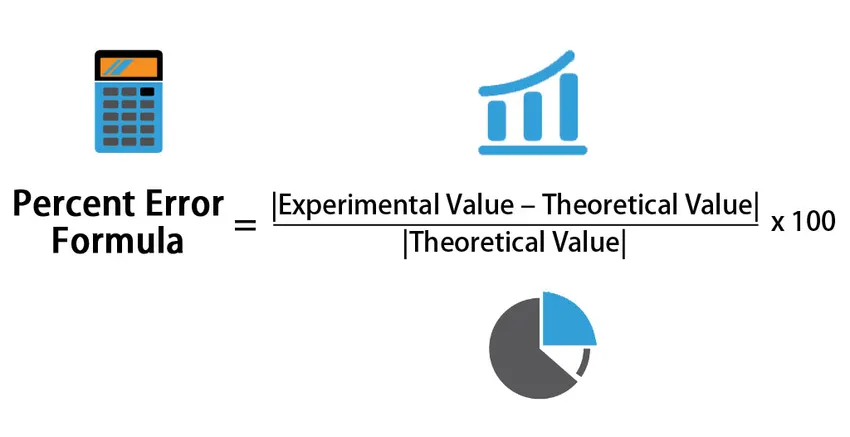
Формула для Процентной Ошибки —
Percent Error = |(Experimental Value – Theoretical Value)| / |Theoretical Value| * 100
Примеры формулы ошибки процента (с шаблоном Excel)
Давайте рассмотрим пример, чтобы лучше понять вычисление Процентной ошибки.
Вы можете скачать этот шаблон ошибок процентов здесь — Шаблон ошибок процентов
Пример № 1
Плотность алюминиевого блока составляет 2, 68 г / см куб., А плотность того же алюминиевого блока при комнатной температуре должна составлять 2, 70 г / см куб. Рассчитать процентную погрешность измерения.
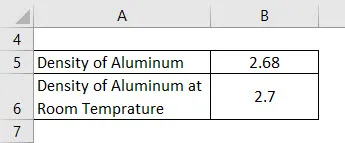
Решение:
Процент ошибок рассчитывается по формуле, приведенной ниже
Процентная ошибка = | (Экспериментальное значение — теоретическое значение) | / | Теоретическая ценность | * 100
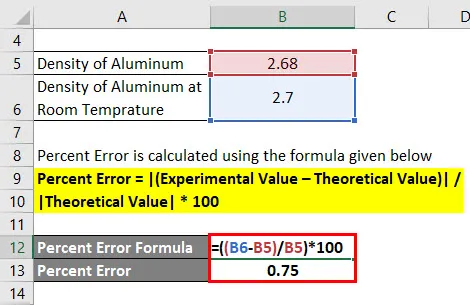
- Процентная ошибка = | (2, 7 — 2, 68) | / | 2, 68 | * 100
- Процентная ошибка = 0, 75%
Пример № 2
Пользователь должен найти объем и массу куба бумаги в экспериментальной лаборатории. Когда пользователь вычисляет значение измерений, он получает значение в виде куба 8, 78 г / см. Принимая во внимание, что принятая плотность куба должна быть 8, 96 г / см куб. Рассчитать процентную ошибку.
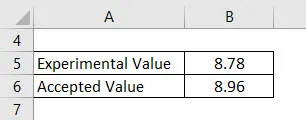
Решение:
Процент ошибок рассчитывается по формуле, приведенной ниже
Процентная ошибка = | (Экспериментальное значение — теоретическое значение) | / | Теоретическая ценность | * 100
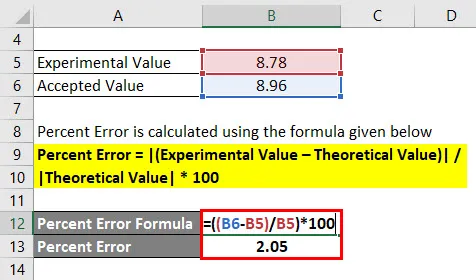
- Процентная ошибка = | (8, 96 — 8, 78) | / | 8, 78 | * 100
- Процент ошибок = 2, 05%
Пример № 3
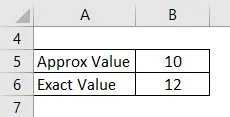
Шкала неверно измеряет значение 10 см из-за некоторой предельной ошибки в вычислениях. Пользователь должен рассчитать процентную погрешность измерения, когда фактическое значение шкалы должно быть 12 см.
Решение:
Процент ошибок рассчитывается по формуле, приведенной ниже
Процентная ошибка = | (Экспериментальное значение — теоретическое значение) | / | Теоретическая ценность | * 100
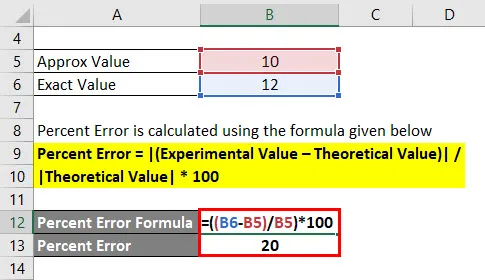
- Процентная ошибка = | (12 — 10) | / | 10 | * 100
- Процент ошибок = 20%
объяснение
Шаги для расчета формулы:
- Вычесть одно значение из другого значения, так как знак формулы не имеет значения, поэтому любое значение можно вычесть из любого значения. Результирующее значение — это значение вашей ошибки, которое просто вычисляется путем вычитания одного значения из другого значения.
- Разделите значение ошибки, которое вычисляется на точное значение или теоретическое значение, которое затем приводит к десятичному числу.
- После вычисления десятичное значение просто преобразует любое десятичное число, вычисленное в процент, умножив его на 100.
Формула покажет значение в процентах.
Релевантность и использование формулы процента ошибок
- Ключом к правильному сообщению процентной ошибки является то, чтобы знать, нужно ли сбрасывать знак (положительный или отрицательный) в расчете, и сообщать значение, используя правильное количество значащих цифр.
- Он часто используется в анализе научных исследований и используется врачом и в области химии. Студенты также, когда используют для проведения экспериментов в лабораториях, используют формулу процента ошибок, чтобы определить отклонение. Приемлемый диапазон ошибок зависит от конкретного случая, и квант процента ошибки должен быть определен заранее, всегда проводя эксперимент с определенным тестом в лаборатории. Использование значения с высокой процентной погрешностью в измерении является суждением об использовании.
Калькулятор формулы ошибки процента
Вы можете использовать следующий калькулятор формулы процента ошибок
| Экспериментальная ценность | |
| Теоретическая ценность | |
| Процент ошибок | |
| Процентная ошибка = |
|
||||||||||
|
Рекомендуемые статьи
Это было руководство к формуле процента ошибок. Здесь мы обсудим, как рассчитать процент ошибок, а также на практических примерах. Мы также предоставляем калькулятор процентов ошибок с загружаемым шаблоном Excel. Вы также можете посмотреть следующие статьи, чтобы узнать больше —
- Руководство по суточной формуле сложных процентов
- Как рассчитать зарплату по формуле?
- Формула для соотношения рынка к книге
- Примеры формул стоимости погашения
|
|

Макеты страниц
Индикаторная ошибка. Изменение цвета индикатора происходит не абсолютно точно в точке эквивалентности, а раньше или позже, поэтому химик-аналитик допускает некоторую ошибку, прекращая титрование раньше или позже требуемого момента. Такую ошибку называют индикаторной ошибкой. Величина ее может колебаться в самых широких пределах в зависимости от того, насколько удачно выбран примененный индикатор.
Предположим, что для титрования 0,1 н. раствора слабой кислоты с  н. раствором сильной щелочи мы использовали два индикатора: метиловый оранжевый
н. раствором сильной щелочи мы использовали два индикатора: метиловый оранжевый  и фенолфталеин
и фенолфталеин  . Посмотрим, что произойдет в обоих случаях титрования.
. Посмотрим, что произойдет в обоих случаях титрования.  исходной кислоты равен 2,87; в точке эквивалентности
исходной кислоты равен 2,87; в точке эквивалентности  (см. § 16).
(см. § 16).
В случае прибавления к титруемой кислоте метилового оранжевого раствор окрасится в красный цвет. Вскоре после прибавления первых порций сильного основания  раствора станет равным 3,1 и раствор окрасится в оранжевый цвет, а при
раствора станет равным 3,1 и раствор окрасится в оранжевый цвет, а при  раствор окрасится в желтый цвет, не изменяющийся при дальнейшем добавлении щелочи. Следовательно, после прибавления нескольких миллилитров, а быть может и капель щелочи придется прекратить титрование раньше достижения точки эквивалентности. Рассчитанное количество титруемой кислоты может оказаться в несколько раз ниже, чем действительное ее содержание, и индикаторная ошибка может достичь 75—85%.
раствор окрасится в желтый цвет, не изменяющийся при дальнейшем добавлении щелочи. Следовательно, после прибавления нескольких миллилитров, а быть может и капель щелочи придется прекратить титрование раньше достижения точки эквивалентности. Рассчитанное количество титруемой кислоты может оказаться в несколько раз ниже, чем действительное ее содержание, и индикаторная ошибка может достичь 75—85%.
Если же к титруемой кислоте прибавить фенолфталеин, то раствор будет оставаться бесцветным до тех пор, пока по мере прибавления к нему щелочи  раствора не достигнет 8,0. При
раствора не достигнет 8,0. При  раствор порозовеет, а при
раствор порозовеет, а при  раствор окрасится в красный цвет.
раствор окрасится в красный цвет.
Так как в точке эквивалентности  что укладывается в интервал
что укладывается в интервал  , то рассчитанное количество кислоты будет практически отвечать действительному ее содержанию и индикаторная ошибка составит
, то рассчитанное количество кислоты будет практически отвечать действительному ее содержанию и индикаторная ошибка составит  .
.
Таким образом, если индикатор выбран правильно, то индикаторную ошибку не принимают во внимание; если же индикатор выбран неправильно, то индикаторная ошибка превышает допустимые погрешности и может достигать очень большой величины. В случаях, требующих более точных результатов, следует учитывать индикаторные ошибки.
Типы индикаторных ошибок. К индикаторным ошибкам относят такие, которые вызываются недотитрованием или перетитрованием исследуемого раствора. В методе нейтрализации различают несколько типов индикаторных ошибок.
а) Водородная ошибка титрования, вызываемая наличием в титруемом растворе по окончании титрования избытка ионов водорода, остающихся в растворе в результате недотитрования сильной кислоты сильной щелочью (обозначается  -ошибка) или перетитрования сильного основания сильной кислотой (обозначается
-ошибка) или перетитрования сильного основания сильной кислотой (обозначается  -ошибка).
-ошибка).
б) Гидроксильная ошибка титрования, вызываемая наличием в титруемом растворе по окончании титрования избытка ионов гидроксила, остающихся в растворе в результате недотитрования сильного основания сильной кислотой ( -ошибка) или перетитрования сильной кислоты сильной щелочью (
-ошибка) или перетитрования сильной кислоты сильной щелочью ( -ошибка).
-ошибка).
в) Кислотная ошибка титрования, вызываемая присутствием в титруемом растворе по окончании титрования нейтральных молекул недотитрованной слабой кислоты ( -ошибка).
-ошибка).
г) Щелочная ошибка титрования, вызываемая присутствием в титруемом растворе по окончании титрования нейтральных молекул недотитрованного слабого основания ( -ошибка).
-ошибка).
Примеры вычисления ошибок титрования.  шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с
шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с  (например, метиловый оранжевый). Титрование заканчивается при
(например, метиловый оранжевый). Титрование заканчивается при  , т. е. при
, т. е. при  (в кислой среде). Следовательно, часть титруемой кислоты будет недотитрована и мы допустим
(в кислой среде). Следовательно, часть титруемой кислоты будет недотитрована и мы допустим  -ошибку. Вычислим ее величину.
-ошибку. Вычислим ее величину.
Пусть концентрация титруемой кислоты  начальный объем кислоты
начальный объем кислоты  объем раствора в конце титрования
объем раствора в конце титрования  .
.
Каждый миллилитр 0,1 н. раствора кислоты или щелочи содержит  г-экв. Для
г-экв. Для  титрования взято
титрования взято  г-экв кислоты.
г-экв кислоты.
Неоттитрованных ионов водорода  останется
останется  , или
, или  раствора
раствора  г-экв кислоты. Эта величина и составляет водородную ошибку титрования, обусловливаемую недотитрованием
г-экв кислоты. Эта величина и составляет водородную ошибку титрования, обусловливаемую недотитрованием  -ионов.
-ионов.
Величину  -ошибки (в процентах) вычисляют согласно пропорции:
-ошибки (в процентах) вычисляют согласно пропорции:  ошибка
ошибка 

В нашем примере

 -шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с
-шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с  (например, фенолфталеин). В этом случае титрование заканчивается при
(например, фенолфталеин). В этом случае титрование заканчивается при  , т. е. при
, т. е. при  щелочной среде. Следовательно, при титровании будет прилит некоторый избыток щелочи, что приведет к
щелочной среде. Следовательно, при титровании будет прилит некоторый избыток щелочи, что приведет к  -ошибке. Вычислим ее величину.
-ошибке. Вычислим ее величину.
Концентрация щелочи к концу титрования составит:

или

В  раствора
раствора  г-экв щелочи.
г-экв щелочи.
Эта величина и составляет гидроксильную ошибку титрования, обусловливаемую перетитрованием кислоты щелочью.
В процентах:

В нашем примере (при  :
:

Это означает, что при титровании сильной кислоты сильным основанием в присутствии метилового оранжевого изменение окраски индикатора наступает раньше, а при титровании в присутствии фенолфталеина (при условии отсутствия в растворе  ) — после достижения точки эквивалентности, причем ошибка титрования при использовании фенолфталеина в 10 раз меньше ошибки титрования с индикатором метиловым оранжевым.
) — после достижения точки эквивалентности, причем ошибка титрования при использовании фенолфталеина в 10 раз меньше ошибки титрования с индикатором метиловым оранжевым.
Так как величина концентрации титруемого раствора входит в знаменатель дроби, то ошибка титрования будет тем больше, чем менее концентрированный раствор титруют.
Поэтому, чтобы избежать больших ошибок титрования, не следует титровать слишком разбавленные растворы очень разбавленными титрантами.
Влияние области интервала перехода индикатора на величину ошибки титрования. При титровании оснований в присутствии индикатора метилового оранжевого  необходимо не только прибавить требуемое по расчету количество сильной кислоты, но и добавить некоторый ее избыток, для того чтобы окраска индикатора изменилась от желтой к оранжево-красной.
необходимо не только прибавить требуемое по расчету количество сильной кислоты, но и добавить некоторый ее избыток, для того чтобы окраска индикатора изменилась от желтой к оранжево-красной.
Если  , то этот избыток (
, то этот избыток ( ) вычисляют по формуле:
) вычисляют по формуле:

При  эта величина возрастает до
эта величина возрастает до  . Такая ошибка совершенно недопустима. Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании оснований индикаторы, интервалы перехода которых лежат ниже
. Такая ошибка совершенно недопустима. Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании оснований индикаторы, интервалы перехода которых лежат ниже  .
.
При титровании кислоты в присутствии фенолфталеина  необходимо прибавить некоторый избыток щелочи, чтобы индикатор из бесцветной формы перешел в окрашенную. Избыток
необходимо прибавить некоторый избыток щелочи, чтобы индикатор из бесцветной формы перешел в окрашенную. Избыток  находят по формуле:
находят по формуле:

При  , равном 11 и 12, эта величина возрастает до 0,5 и
, равном 11 и 12, эта величина возрастает до 0,5 и  . Такая ошибка совершенно недопустима.
. Такая ошибка совершенно недопустима.
Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании кислот индикаторы, интервалы перехода которых лежат выше  .
.
Гидроксильная ошибка титрования. Предположим, что для титрования сильного основания сильной кислотой выбран индикатор с  концентрация титруемого основания
концентрация титруемого основания  начальный объем основания
начальный объем основания  объем в конце титрования
объем в конце титрования  .
.
Каждый миллилитр 0,1 н. раствора основания содержит г-экв.
Для титрования взято  г-экв основания. Титрование заканчивают при
г-экв основания. Титрование заканчивают при  (в щелочной среде); следовательно,
(в щелочной среде); следовательно,  , т. е.
, т. е.  . Таким образом, часть титруемой щелочи будет недотитрована, и мы допустим
. Таким образом, часть титруемой щелочи будет недотитрована, и мы допустим  -ошибку. Вычислим ее величину.
-ошибку. Вычислим ее величину.
Концентрация ионов гидроксила в конце титрования составит  или
или  .
.
В  раствора
раствора  основания.
основания.
Эта величина и составляет гидроксильную ошибку титрования, обусловленную недотитрованием ОН « -ионов.
-ионов.
Величину  -ошибки (в процентах) вычисляют по формуле:
-ошибки (в процентах) вычисляют по формуле:

В нашем примере

Если выполнять титрование в присутствии индикатора с  , то необходимо прибавить некоторый избыток кислоты.
, то необходимо прибавить некоторый избыток кислоты.
Водородная ошибка титрования. Предположим, что для титрования сильного основания сильной кислотой выбран индикатор  . Титрование заканчивают при
. Титрование заканчивают при  — составляет водородную ошибку титрования, обусловленную перетитрованием щелочи кислотой.
— составляет водородную ошибку титрования, обусловленную перетитрованием щелочи кислотой.
В процентах

 — ошибка
— ошибка  (57)
(57)
Если  , то в этом случае получим:
, то в этом случае получим:

Кислотная ошибка титрования. Предположим, что для титрования дана слабая кислота  . Титрование ведут в присутствии индикатора с
. Титрование ведут в присутствии индикатора с  . В этом случае
. В этом случае

или

Так как  для слабого электролита равна
для слабого электролита равна  , то можно написать:
, то можно написать:

 Титрование заканчивается при
Титрование заканчивается при  , следовательно,
, следовательно,  -ошибка равна:
-ошибка равна:

Отношение можно рассматривать как отношение концентраций неоттитрованной части кислоты к оттитрованной ее части и считать это отношение критерием кислотной ошибки титрования. В нашем примере  -ошибка
-ошибка  или
или  . При титровании той же самой кислоты в присутствии индикатора с
. При титровании той же самой кислоты в присутствии индикатора с  соответствующая ошибка увеличилась бы в
соответствующая ошибка увеличилась бы в  :
:

Пример. Вычислите ошибку титрования 0,1 н. раствора  0,1 н. раствором
0,1 н. раствором  в присутствии индикатора метилового оранжевого
в присутствии индикатора метилового оранжевого  :
:

Следовательно, на каждую оттитрованную молекулу  приходится 5,5 неоттитрованных.
приходится 5,5 неоттитрованных.

Это значит, что 85% кислоты будет не оттитровано, если титровать  раствором
раствором  в присутствии метилового оранжевого. Следовательно, титровать
в присутствии метилового оранжевого. Следовательно, титровать  с индикаторами, имеющими
с индикаторами, имеющими  , нельзя.
, нельзя.
Щелочная ошибка титрования. Предположив, что для титрования дано слабое основание КЮН, щелочную ошибку титрования вычисляют аналогично кислотной ошибке:
 (59)
(59)
Пример. Вычислите ошибку титрования 0,1 н. раствора аммиака  0,1 н. раствором
0,1 н. раствором  в присутствии индикатора фенолфталеина
в присутствии индикатора фенолфталеина  :
:

Следовательно, на каждую оттитрованную молекулу  приходится 0,55 неоттитрованной:
приходится 0,55 неоттитрованной:

Это значит, что около 35% аммиака будет не оттитровано, если его титровать раствором  в присутствии фенолфталеина. Следовательно, титровать раствор
в присутствии фенолфталеина. Следовательно, титровать раствор  с индикатором, имеющим
с индикатором, имеющим  , нельзя.
, нельзя.
Оглавление
- ПРЕДИСЛОВИЕ К ПЕРВОМУ ИЗДАНИЮ
- ПРЕДИСЛОВИЕ КО ВТОРОМУ ИЗДАНИЮ
- ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
- ВВЕДЕНИЕ
- § 1. Понятие о количественном анализе
- § 2. Классификация методов количественного анализа
- § 3. Характеристика методов количественного анализа
- § 4. Анализ больших и малых количеств вещества
- § 5. Отбор средней пробы
- § 6. Подготовка вещества для взвешивания
- § 7. Взвешивание
- § 8. Техника взвешивания на аналитических весах
- § 9. Правила обращения с аналитическими весами
- § 10. Приготовление раствора для анализа
- § 11. Запись результатов анализа
- Часть первая. Объемный анализ
- § 1. Сущность объемного анализа
- § 2. Общее уравнение реакции титрования и выводы из него
- Б. ТЕХНИКА ХИМИЧЕСКОГО ЭКСПЕРИМЕНТА В ОБЪЕМНОМ АНАЛИЗЕ
- § 3. Измерение объемов растворов
- § 4. Посуда, применяемая для измерения объемов растворов
- § 5. Работа с мерными колбами
- § 6. Работа с пипетками
- § 7. Работа с бюретками
- § 8. Приготовление стандартных растворов
- В. ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
- § 9. Концентрация растворов и способы ее выражения
- § 10. Способы вычисления в объемном анализе
- § 11. Связь между точностью измерений и точностью вычислений
- § 12. Краткие сведения о статистической обработке экспериментальных данных
- Г. ПОЛУМИКРООБЪЕМНЫЙ МЕТОД АНАЛИЗА
- § 13. Понятие о полумикрообъемном анализе
- § 14. Особенности техники измерения объемов растворов в полумикрометоде
- Д. БЕЗБЮРЕТОЧНЫЕ МЕТОДЫ ТИТРОВАНИЯ
- § 15. Понятие о безбюреточных методах титрования
- § 16. Классификация методов безбюреточного титрования
- Е. АВТОМАТИЧЕСКИЕ МЕТОДЫ
- § 17. Химико-аналитический контроль производства
- § 18. Автоматические методы титрования
- ГЛАВА II. МЕТОДЫ НЕЙТРАЛИЗАЦИИ, ИЛИ МЕТОДЫ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
- § 1. Характеристика метода
- § 2. Установление точки эквивалентности
- § 3. Графический метод изображения процесса нейтрализации
- § 4. Вычисление концентрации ионов водорода в водных растворах сильных кислот и оснований
- § 5. Вычисление активности ионов водорода в водных растворах сильных кислот и оснований
- § 6. Титрование сильной кислоты сильным основанием
- § 7. Вычисление концентрации ионов водорода в растворах слабых кислот и оснований
- § 8. Вычисление активности ионов водорода в водных растворах слабых кислот и оснований
- § 9. Равновесия в водных буферных растворах слабых кислот в присутствии солей этих кислот
- § 10. Равновесия в водных буферных растворах слабых оснований в присутствии солей этих оснований
- § 11. Вычисление концентрации ионов водорода в водных буферных растворах
- § 12. Вычисление активности ионов водорода в водных буферных растворах
- § 13. Вычисление концентрации ионов водорода и степени гидролиза в водных растворах гидролизующихся бинарных солей
- § 14. Вычисление активности ионов водорода в водных растворах гидролизующихся бинарных солей
- § 15. Титрование слабой кислоты сильным основанием
- § 16. Титрование слабого основания сильной кислотой
- § 17. Титрование многоосновных кислот
- § 18. Титрование солей, образованных катионами сильных оснований и анионами слабых многоосновных кислот
- § 19. Изменение активности и показателя активности ионов водорода в процессе титрования водных растворов кислот и оснований
- § 20. Выводы, вытекающие из рассмотрения кривых нейтрализации
- § 21. Индикаторы
- § 22. Интервал перехода индикатора
- § 23. Выбор индикатора
- § 24. Ошибки титрования
- Б. ПРАКТИЧЕСКАЯ ЧАСТЬ
- § 25. Организация рабочего места
- § 26. Приготовление стандартных (титрованных) растворов
- § 27. Приготовление 0,1 н. раствора хлористоводородной кислоты
- § 28. Установка титра 0,1 н. раствора хлористоводородной кислоты
- § 29. Приготовление 0,1 н. раствора едкого натра
- § 30. Установка титра 0,1 н. раствора едкого натра
- § 31. Определение карбонатов
- § 32. Определение содержания H2SO4 в технической серной кислоте
- § 33. Определение содержания уксусной кислоты
- § 34. Определение содержания Na2CO3 и NaOH при их совместном присутствии
- § 35. Определение содержания Na2CO3 и NaHCO3 при их совместном присутствии
- § 36. Определение жесткости воды
- § 37. Определение аммонийного азота в солях аммония
- § 38. Определение содержания фосфорной кислоты
- В. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ В НЕВОДНЫХ СРЕДАХ
- § 39. Неводные растворы
- § 40. Современные представления о кислотах и основаниях
- § 41. Диссоциация электролитов в неводных растворах
- § 42. Влияние неводных растворителей на силу кислот и оснований
- § 43. Применение закона действия масс к растворам сильных электролитов
- § 44. Титрование кислот и оснований в неводных растворах
- § 45. Методы кислотно-основного титрования в неводных средах
- § 46. Примеры практических определений в неводных растворах
- ГЛАВА III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ (ОКСИДИМЕТРИЯ, ОКСРЕДМЕТРИЯ, РЕД-ОКС-МЕТОДЫ)
- § 1. Значение окислительно-восстановительных потенциалов
- § 2. Реакции окисления—восстановления и комплексообразования
- § 3. Примеры окислительно-восстановительного титрования
- § 4. Константы равновесия окислительно-восстановительных реакций
- § 5. Связь между константами равновесия окислительно-восстановительных реакций и нормальными потенциалами
- § 6. Вычисление констант равновесия окислительно-восстановительных реакций
- § 8. Зависимость скорости реакций окисления—восстановления от различных факторов
- § 9. Графический метод изображения процесса окисления—восстановления
- § 10. Фиксирование точки эквивалентности в методах окисления—восстановления
- § 11. Окислительно-восстановительные индикаторы (ред-окс-индикаторы)
- Б. ПЕРМАНГАНАТОМЕТРИЯ
- § 12. Основы перманганатометрии
- § 13. Титрование перманганатом в кислой среде
- § 14. Титрование перманганатом в щелочной среде
- § 15. Приготовление стандартного (титрованного) раствора перманганата калия
- § 16. Установка титра стандартного раствора перманганата калия
- § 17. Установка титра и нормальности раствора перманганата калия по оксалату аммония
- § 18. Вещества, определяемые методом перманганатометрии
- ОПРЕДЕЛЕНИЕ ВОССТАНОВИТЕЛЕЙ
- § 19. Определение щавелевой кислоты и оксалатов
- § 20. Определение соединений железа (II)
- § 21. Определение содержания металлического железа в присутствии окислов железа
- § 22. Определение азотистой кислоты и нитритов
- § 23. Определение содержания марганца (II) в рудах
- ОПРЕДЕЛЕНИЕ ОКИСЛИТЕЛЕЙ
- § 24. Определение соединений железа (III)
- § 25. Определение нитратов
- § 26. Определение бихроматов
- § 27. Определение содержания MnO2 в пиролюзите
- ОПРЕДЕЛЕНИЕ ДРУГИХ ВЕЩЕСТВ
- § 28. Определение ионов кальция
- В. ИОДОМЕТРИЯ
- § 29. Основы иодометрии
- § 30. Методы иодометрического титрования
- § 31. Преимущества и недостатки иодометрического метода
- § 32. Приготовление стандартного (титрованного) раствора тиосульфата и установка его титра
- § 33. Приготовление стандартного (титрованного) раствора иода и установка его титра
- МЕТОДЫ ПРЯМОГО ТИТРОВАНИЯ
- § 34. Определение мышьяка (III)
- МЕТОДЫ ОБРАТНОГО ТИТРОВАНИЯ
- § 35. Определение сульфита натрия
- § 36. Определение содержания формальдегида в формалине
- МЕТОДЫ КОСВЕННОГО ОПРЕДЕЛЕНИЯ
- § 37. Определение ионов меди (II)
- § 38. Определение двуокиси свинца в сурике
- МЕТОД ТИТРОВАНИЯ ЗАМЕСТИТЕЛЕЙ
- § 39. Определение содержания двуокиси марганца в пиролюзите
- МЕТОД ОПРЕДЕЛЕНИЯ КИСЛОТ
- § 40. Определение хлористоводородной кислоты
- § 41. Определение воды по Фишеру
- Г. ПОНЯТИЕ О ДРУГИХ МЕТОДАХ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
- § 42. Хроматометрия
- § 43. Определение содержания железа (II)
- § 44. Цериметрия
- § 45. Броматометрия
- § 46. Ванадатометрия
- § 47. Аскорбинометрия
- § 48. Титанометрия
- ГЛАВА IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
- § 1. Общая характеристика методов
- § 2. Классификация методов осаждения и комплексообразования
- § 3. Применение теории осаждения к объемному анализу
- § 4. Вычисление растворимости электролитов в воде с учетом коэффициентов активности
- § 5. Влияние одноименных ионов на растворимость малорастворимого электролита
- § 6. Солевой эффект
- § 7. Влияние концентрации ионов водорода на растворимость малорастворимых соединений
- § 8. Кривые титрования в методе осаждения
- § 9. Общие выводы, вытекающие из рассмотрения кривых осаждения
- § 10. Адсорбционные явления, наблюдаемые при титровании по методу осаждения
- Б. АРГЕНТОМЕТРИЯ
- § 11. Характеристика метода
- § 12. Приготовление 0,1 н. раствора нитрата серебра
- § 13. Приготовление стандартного раствора хлорида натрия
- § 14. Установка титра 0,1 н. раствора нитрата серебра по точной навеске хлорида натрия
- §15. Определение ионов хлора в техническом хлориде натрия по методу Мора
- § 16. Определение хлоридов по методу Фаянса
- В. РОДАНОМЕТРИЯ
- § 17. Характеристика метода
- § 18. Приготовление 0,1 н. раствора роданида аммония
- § 19. Определение ионов хлора в растворимых хлоридах по методу Фольгарда
- § 20. Определение серебра в сплавах
- Г. МЕРКУРИМЕТРИЯ
- § 21. Характеристика метода
- § 22. Приготовление 0,1 н. раствора нитрата ртути (II)
- § 23. Установка титра раствора нитрата ртути (II)
- § 24. Определение ионов хлора в воде меркуриметрическим методом
- Д. МЕРКУРОМЕТРИЯ
- § 25. Краткая характеристика метода
- Е. КОМПЛЕКСОНОМЕТРИЯ (ХЕЛАТОМЕТРИЯ)
- § 26. Характеристика метода
- § 27. Теоретические основы комплексонометрического титрования
- § 28. Классификация методов комплексонометрического титрования
- § 29. Установка титра раствора комплексона III
- § 30. Определение содержания кальция
- § 31. Определение жесткости воды комплексонометрическим методом
- § 32. Анализ смеси ионов кальция и магния
- § 33. Определение содержания алюминия
- § 34. Раздельное определение ионов кальция и алюминия
- § 35. Раздельное определение ионов алюминия и железа
- Часть вторая. Весовой анализ
- § 1. Сущность весового анализа
- § 2. Классификация методов весового анализа
- § 3. Расчеты в весовом анализе
- Б. ТЕХНИКА ВЕСОВОГО АНАЛИЗА
- § 4. Взятие и растворение навески
- § 5. Техника осаждения
- § 6. Фильтрование и промывание осадков
- § 7. Получение весовой формы
- § 8. Взвешивание весовой формы
- В. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
- § 9. Теоретические основы выделения осадков из растворов с помощью специфических неорганических и органических реактивов
- § 10. Требования, предъявляемые к осадкам
- § 11. Методы повышения точности весовых определений
- § 12. Теоретические обоснования выбора оптимальных условий для весового определения
- Г. ПРАКТИЧЕСКАЯ ЧАСТЬ
- § 13. Определение кристаллизационной воды в BaCl2 2H2O
- § 14. Определение сульфат-ионов или серы
- § 15. Определение ионов железа (III)
- § 16. Определение содержания кальция в карбонате кальция
- § 17. Определение содержания магния
- § 18. Определение ионов хлора в растворимых хлоридах или в хлористоводородной кислоте
- § 19. Анализ силикатов
- § 20. Анализ доломита
- § 21. Анализ бронзы и латуни
- Д. МЕТОДЫ ВЕСОВЫХ ОПРЕДЕЛЕНИЙ, ОСНОВАННЫЕ НА ПРИМЕНЕНИИ ОРГАНИЧЕСКИХ РЕАКТИВОВ
- § 22. Определение никеля
- § 23. Определение алюминия
- Часть третья. Понятие о физических и физико-химических (инструментальных) методах анализа
- § 1. Электрохимические методы
- § 2. Спектральные (оптические) методы
- § 3. Хроматографические методы
- § 4. Радиометрические методы
- § 5. Масс-спектрометрические методы
- ГЛАВА VII ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
- § 1. Характеристика методов электроанализа
- § 2. Химические процессы, протекающие при электролизе
- § 3. Методы электроанализа
- § 4. Электровесовой анализ
- § 5. Метод внутреннего электролиза
- § 6. Определение меди в растворе сульфата меди с применением платиновых сетчатых электродов
- § 7. Определение меди и свинца в латуни с применением платиновых сетчатых электродов
- § 8. Определение малых количеств меди методом внутреннего электролиза
- ГЛАВА VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
- § 1. Особенности объемных электрохимических методов анализа
- § 2. Кондуктометрическое титрование
- § 3. Высокочастотное титрование
- § 4. Потенциометрическое титрование
- § 5. Полярографический метод анализа
- § 6. Амперометрическоб титрование
- § 7. Кулонометрическое титрование
- ГЛАВА IX. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
- § 1. Понятие об эмиссионном спектральном анализе
- Б. КОЛОРИМЕТРИЯ
- § 2. Особенности колориметрических методов анализа
- § 3. Характеристика колориметрических методов анализа
- В. ОПТИЧЕСКИЕ МЕТОДЫ УСТАНОВЛЕНИЯ ТОЧКИ ЭКВИВАЛЕНТНОСТИ
- § 4. Спектрофотометрическое титрование
- § 5. Фототурбидиметрическое и фотонефелометрическое титрование
- Г. ЛАБОРАТОРНЫЕ РАБОТЫ
- § 6. Определение содержания ионов железа методом колориметрического титрования
- § 7. Определение содержания титана
- ГЛАВА X. МЕТОДЫ РАЗДЕЛЕНИЯ, ВЫДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ ОТДЕЛЬНЫХ КОМПОНЕНТОВ АНАЛИЗИРУЕМЫХ СМЕСЕЙ
- § 1. Определение следов элементов (микропримесей)
- § 2. Метод осаждения малорастворимых соединений
- § 3. Электрохимические методы разделения
- § 4. Метод экстрагирования
- § 5. Методы отгонки летучих соединений
- § 6. Хроматографические методы разделения
- § 7. Метод флотации
Для
выявления ошибок и их численной оценки
(особенно при разработке новых
аналитических методик) количественный
анализ повторяют несколько раз, т.е.
проводят параллельные
определения. Под
параллельными определениями понимают
получение нескольких результатов
единичных определений для одной и той
же пробы в одинаковых условиях.
Пусть
μ
— истинное
значение определяемой величины; Х1,
Х2,
…, Хi,
…, …, Хn
— измеренные
(единичные) значения определяемой
величины — результаты единичных
определений; п- общее
число единичных определений.
Под
единичным определением понимают
однократное проведение всей
последовательности операций,
предусмотренных методикой анализа.
Результат единичного определения- это
значение содержания определяемого
компонента, найденное при единичном
определении.
Иногда
(часто) вместо истинного значения
определяемой величины μ
используют действительное
значение содержания а
(или просто
действительное
значение а),
под которым
подразумевают экспериментально
полученное или расчетное значение
определяемого содержания, настолько
близкое к истинному, что для данной цели
может быть использовано вместо него.
Тогда величина

есть
среднее
арифметическое (среднее) из
результатов единичных определений.
Считается, что х–
— наиболее вероятное значение определяемой
величины, более вероятное, чем каждое
отдельное значение Хi.
Под
правильностью
результата
анализа понимают качество анализа,
отражающее близость к нулю разности
между средним арифметическим и истинным
μ
(или действительным а)
значением определяемой величины:

Другими
словами, правильность результата анализа
отражает близость полученного среднего
значения х–
к истинному (или действительному)
значению определяемой величины.
Воспроизводимость
результата
анализа характеризует степень близости
результатов единичных определений Хi,
друг к другу.
Правильность
и воспроизводимость результата анализа
зависят от различного типа ошибок.
1.4.2. Классификация ошибок количественного анализа.
Ошибки
количественного анализа условно
подразделяют
на систематические,
случайные и грубые.
Грубые
ошибки,
обусловленные
несоблюдением методики анализа, очевидны.
Они устраняются при повторном проведении
анализа с соблюдением всех требуемых
условий, предусмотренных методикой
анализа.
а)
Систематическая ошибка
Различают:
систематическую
ошибку и
процентную
систематическую ошибку.
Систематическая
ошибка результата
анализа Δ0
— это статистически значимая разность
между средним х–
и действительным а
(или истинным
μ)
значениями содержания компонента:

Систематическая
ошибка результата анализа может быть
больше нуля, меньше нуля или равна нулю.
Процентная
систематическая ошибка (относительная
величина систематической ошибки) —
это систематическая ошибка, выраженная
в процентах от действительного значения
а
(или истинного
значения μ)
определяемой величины:
δ
= (х–
– а)∙100%
/ а
или δ = (х–
– μ)•100% / μ (1.3)
Для
относительной величины систематической
ошибки вместо символа 5 используют
также обозначение Δ0
%.
Систематическая
ошибка характеризует правильность
результатов анализа; поэтому правильность
анализа можно определить так же, как
качество анализа, отражающее близость
к нулю систематической ошибки.
Систематические
ошибки обусловлены либо постоянно
действующими причинами (и поэтому они
повторяются при многократном проведении
анализа), либо изменяются по постоянно
действующему закону, который можно
учесть.
Так,
например, процентная систематическая
ошибка (Δс/с)•100% фотометрических
определений (с
— концентрация,
Δс — систематическая ошибка определения
концентрации фотометрическим методом)
минимальна в интервале изменений
оптической плотности А
от А
≈ 0,2 до А
≈ 0,8 и
составляет (Δс/с)•100% < 0,4%.
Источники
систематических ошибок. Невозможно
с исчерпывающей полнотой перечислить
все источники систематических ошибок.
Основными являются следующие.
Методические
— обусловлены
особенностями методики анализа. Например,
аналитическая реакция прошла не до
конца; имеются потери осадка вследствие
его частичной растворимости в растворе
или при его промывании; наблюдается
соосаждение примесей с осадком, вследствие
чего масса осадка возрастает, и т.д.
Инструментальные
— обусловлены
несовершенством используемых приборов
и оборудования. Так, например,
систематическая ошибка взвешивания на
лабораторных аналитических весах
составляет ±0,0002 г. Систематическая
ошибка в титриметрических методах
анализа вносится вследствие неточности
калибровки бюреток, пипеток, мерных
колб, цилиндров, мензурок и т.д.
Индивидуальные
— обусловлены
субъективными качествами аналитика.
Так, например, дальтонизм может влиять
на определение конечной точки титрования
при визуальной фиксации изменения
окраски индикатора.
В
конечном итоге правильность результатов
анализа определяется наличием или
отсутствием именно систематических
ошибок.
Существуют способы
выявления систематических ошибок.
а)
Использование стандартных, образцов.
Общий состав
стандартного образца должен быть близким
к составу анализируемой пробы, а
содержание определяемого компонента
в стандартном образце должно быть точно
известно.
Анализ
стандартного образца — наиболее надежный
способ выявления наличия или отсутствия
систематической ошибки и оценки
правильности результата анализа.
б) Анализ
исследуемого объекта другими методами.
Исследуемый
объект анализируют методом или методами,
которые не дают систематической ошибки
(метрологически аттестованы), и сравнивают
результаты анализа с данными, полученными
при анализе того же объекта с использованием
оцениваемой методики или не аттестованного
оборудования. Сравнение позволяет
охарактеризовать правильность оцениваемой
методики анализа.
в)
Метод добавок или метод удвоения —
используют при отсутствии стандартных
образцов и метрологически аттестованной
методики (метода) анализа.
Анализируют
образец, используя оцениваемую методику.
Затем удваивают массу анализируемой
пробы или увеличивают (уменьшают) массу
в иное число раз, снова находят содержание
определяемого компонента в уже новой
пробе и сравнивают результаты анализов.
Должна выполняться определенная
закономерность (например, пропорциональность).
б)
Случайные ошибки.
Случайные
ошибки показывают отличие результатов
параллельных определений друг от друга
и фактически характеризуют воспроизводимость
анализа. Причины
случайных ошибок однозначно указать
невозможно. При многократном повторении
анализа они или не воспроизводятся, или
имеют разные численные значения и даже
разные знаки.
Случайные
ошибки можно оценить методами
математической статистики, если
выявлены и устранены систематические
ошибки (или систематические ошибки
меньше случайных).
ЭТАПЫ
КОЛИЧЕСТВЕННОГО ХИМИЧЕСКОГО АНАЛИЗА
Наука
об измерениях, методах и средствах
обеспечения их единства
и способах достижения требуемой точности
называется метрологией.
Количественный
химический анализ, целью которого
является
определение содержания веществ в разных
объектах, может
рассматриваться как измерительная
процедура, характеризующаяся
рядом специфических особенностей.
Количественный
химический анализ, прежде всего, является
многостадийным процессом, включающим
ряд этапов и стадий. При выполнении
химического анализа с помощью любого
метода можно выделить следующие
основные этапы:
— постановка
аналитической задачи;
— выбор метода
анализа;
— выполнение
анализа;
— оценка качества
анализа;
— принятие решений
по результатам анализа.
При
постановке аналитической задачи
необходимо дать характеристику объекта
анализа, указать химическую формулу
определяемого
компонента, возможный интервал его
содержаний, требуемую
точность и продолжительность анализа.
Выбор
метода анализа определяется поставленной
аналитической
задачей и техническими возможностями
аналитической лаборатории.
Этап,
связанный непосредственно с проведением
химического
анализа, наиболее трудоемок и включает
ряд стадий, представленных на рис. 5.2.

Методика
анализа включает
подробное описание последовательности
и условий проведения всех стадий анализа.
Точное следование
методике анализа позволяет выполнить
анализ с минимальными
погрешностями на каждой стадии и получить
правильный
результат анализа.
Первая
стадия химического анализа — отбор
средней
(представительной)
пробы. Это
небольшая часть анализируемого объекта,
средний
состав и свойства которой должны быть
идентичны во всех
отношениях среднему составу и свойствам
объекта анализа. Содержание
определяемого компонента в анализируемой
пробе должно
отражать среднее содержание этого
компонента во всем исследуемом
объекте, т. е. анализируемая проба должна
быть представительной.
Погрешность в отборе пробы часто
определяет общую
погрешность химического анализа. Не
оценив погрешность на
этой стадии, нельзя говорить о правильности
определения компонента
в анализируемом объекте.
Подготовка
пробы к анализу включает ряд сложных
операций, например, такие как высушивание
пробы, разложение (вскрытие)
пробы,
устранение влияния мешающих компонентов.
В зависимости
от цели анализа, природы объекта и
выбранного метода могут быть использованы
разные модификации и комбинации этих
операций.
В правильном проведении химического
анализа роль подготовки
пробы настолько велика, что химик-аналитик
должен каждый
раз оценивать необходимость включения
операций пробоподготовки
в методику анализа, их влияние на общую
погрешность анализа.
После
отбора и подготовки пробы наступают
стадии химического
анализа, на которых определяют количество
компонента. С
этой целью измеряют аналитический
сигнал. В
большинстве методов им является среднее
из измерений физической величины на
заключительной стадии анализа,
функционально связанной с содержанием
определяемого компонента. Это
может быть сила тока, ЭДС системы,
оптическая плотность, интенсивность
излучения и т.д. В отдельных методах
анализа возможно непосредственное
определение содержания. Например, в
гравиметрическом методе иногда прямо
измеряют массу определяемого компонента.
При
определении количества компонента
измеряют величину аналитического
сигнала. Затем рассчитывают содержание
компонента с использованием функциональной
зависимости аналитического
сигнала от содержания: у =
f(с),
которая устанавливается расчетным или
опытным путем и может быть представлена
в виде формулы, таблицы или графика.
Содержание может быть выражено абсолютным
количеством определяемого компонента
в молях, в единицах массы или через
соответствующие концентрации.
При
измерении аналитического сигнала
учитывают наличие полезного аналитического
сигнала, являющегося функцией содержания
определяемого компонента, и аналитического
сигнала фона, обусловленного
примесями определяемого компонента и
мешающими компонентами в растворах,
растворителях и матрице образца, а также
«шумами» в
измерительных приборах, усилителях
и другой аппаратуре. Эти шумы не имеют
отношения к определяемому компоненту,
но накладываются на его собственный
аналитический сигнал. Задача аналитика
состоит в том, чтобы максимально снизить
величину аналитического сигнала фона
и, главное, сделать минимальными его
колебания.
Обычно
аналитический сигнал фона учитывают в
контрольном
(холостом) опыте, когда
через все стадии химического анализа
проводится проба, не содержащая
определяемого компонента. Полезным при
этом будет аналитический сигнал, равный
разности измеренного сигнала и
аналитического сигнала фона.
На
основании существующей зависимости
между аналитическим сигналом и содержанием
находят концентрацию определяемого
компонента. Обычно при этом используют
методы
градуированного графика, стандартов
или
добавок. Описанные
в литературе другие способы определения
содержания компонента, как правило,
являются модификацией этих трех методов.
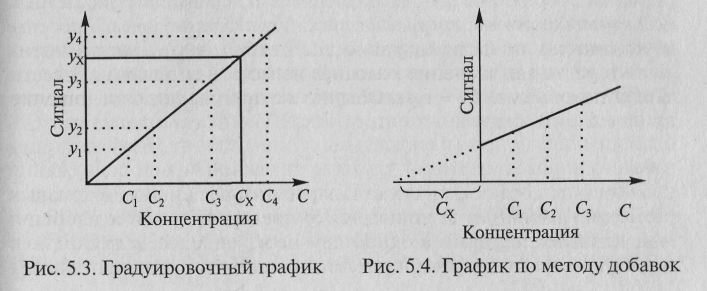
Наиболее
распространен метод
градуировочного графика: в
координатах (аналитический сигнал –
содержание компонента) строят график
с использованием образцов сравнения с
разными и точно известными уровнями
содержания компонента (концентрация
С).
Затем, измерив
величину аналитического сигнала в
пробе, находят содержание определяемого
компонента по градуировочному графику
(рис. 5.3).
В
методе
стандартов измеряют
аналитический сигнал в образце
сравнения (стандартном образце) с
известным содержанием компонента и в
анализируемой пробе: Уст
= sСст
и ух
= sСХ,
где s-коэффициент
пропорциональности.
Если
определенное в идентичных условиях
значение s
заранее известно, то можно провести
расчет по формуле Сх
= Ух/S.
Обычно же используют соотношение Уст/УХ
= Сст/СХ,
откуда
![]()
Иногда
работают с двумя стандартными образцами,
в которых содержание компонента
отличается от предполагаемого содержания
в анализируемой пробе в одном случае в
меньшую, в другом — в большую сторону.
Этот вариант метода стандартов называют
методом
ограничивающих растворов.
Содержание определяемого компонента
рассчитывают по формуле

Если
при определении малых количеств
компонента нужно учесть
влияние матрицы образца на величину
аналитического сигнала,
то часто используют
методы добавок — расчетный
или графический.
При
определении содержания расчетным
методом берут
две аликвоты
раствора анализируемой пробы. В одну
из них вводят добавку
определяемого компонента известного
содержания. В обеих
пробах измеряют аналитический сигнал
— ух
и
ух+доб—
Неизвестную
концентрацию определяемого компонента
рассчитывают
по формуле
![]()
где
Vдоб
и Сдоб
— объем и концентрация добавленного
раствора определяемого компонента; V
— аликвота
анализируемой пробы.
При
определении содержания компонента
графическим
методом
берут
п
аликвот
анализируемой пробы: 1, 2, 3, …, п.
Во
2-ю, …, п-ю
аликвоты
вводят известные, возрастающие, количества
определяемого
компонента. Во всех аликвотах измеряют
аналитический сигнал
и строят график в координатах аналитический
сигнал -содержание
определяемого компонента, приняв за
условный нуль содержание
определяемого компонента в аликвоте
без добавки (аликвота 1). Экстраполяция
полученной прямой до пересечения с
осью абсцисс дает отрезок слева от
условного нуля координат, величина
которого в выбранном масштабе и единицах
измерения соответствует
Сх
определяемого компонента (рис. 5.4).
Метод
стандартов и метод добавок применимы
для линейной градуировочной функции.
Метод градуировочного графика допускает
использование как линейной, так и
нелинейной функций аналитический
сигнал—содержание. В последнем случае
требуется большее число экспериментальных
данных, и результат определения содержания
компонента бывает, как правило, менее
точным.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #